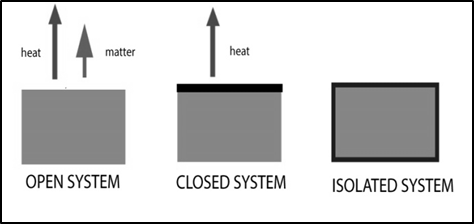
aContents:IntroductionSystem and SurroundingsProperties of the systemThermodynamic processesState function & Path functionInternal energy (U)Heat (q)Work (w)Work involved in expansion and compression processesFirst Law of ThermodynamicsEnthalpy (H)Enthalpy Changes for different types of Reactions and Phase TransitionsThermochemical equationsHeat capacityRelation between Cp and Cv for an ideal gasHess’s law of constant heat summationNeed for the Second law of thermodynamicsSecond Law of thermodynamicsEntropy (S)Gibbs free energy (G)Relationship between standard free energy change (ΔGo) and equilibrium constant (Keq)Helmholtz energyThird law of Thermodynamics6.1 IntroductionIn our daily life, we come across many useful reactions such as flow of electrons through circuit to produce electrical energy, metabolic reactions to produce the energy required for biological functions and so on. Thermodynamics [‘Thermos’ (heat) and `dynamics’ (flow)] is concerned with flow of heat and the relation between heat and work. It deals with heat and its relationship with energy in any one of its different forms; mechanical, chemical, electrical, etc. Only macroscopic observable properties (pressure, temperature, internal energy, etc.) of matter are considered without assumptions of its atomic nature. It is based on three simple Laws that are not derived but are generalizations deduced from long experience of the scientists with energy and of natural phenomena. These laws have been used to deduce powerful mathematical relationships applicable to a broad range of processes. Laws of thermodynamics apply only when a system is in equilibrium or moves from one equilibrium state to another equilibrium state but does not take into account the kinetic approach to the equilibrium state.6.2 System and SurroundingsIn thermodynamics, system is a quantity of matter or part of universe chosen for observation purpose separated by a boundary from the rest of the universe. The system may be water in a beaker, a balloon filled with air, an aqueous solution of glucose etc. All other parts of the universe outside the boundary that is not the part of the system is called surroundings. Boundary is designed to allow us to control and keep track of movements of matter and energy in or out of the system.6.2.1 Types of SystemDifferent types of systems are possible based on the movements of matter and energy in or out of the system.
Open system: A system can exchanged both energy and matter with the surroundings is said to be open system. An open frying pan is an example of open system.Closed system: A system which can exchange only energy (not matter) with the surroundings is said to be closed system. A gas contained in a cylinder fitted with a piston constitutes a closed system.Isolated system: A system which can exchange neither matter nor energy with its surroundings is said to be an isolated system. Here boundary is sealed and insulated. Hot water contained in a thermos flask or any other closed insulated vessel is an example for an isolated system.6.3 Properties of the systemThe experimentally measurable physical properties which enable us to define a system are called thermodynamic parameters or properties of the system. Examples are number of moles, energy, temperature, pressure, volume, density, etc. Some of the properties of a system depend on its mass or size whereas other properties do not depend on its mass or size. Based on this, the properties of a system fall into two classes.Extensive properties: The property that depends on the mass or the size of the system is called an extensive property. Examples are volume, number of moles, energy, surface area, etc.Intensive properties: The property that is independent of the mass or the size of the system is called an intensive property. Examples are surface tension, density, temperature, molar volume, pressure etc.6.4 Thermodynamic processesThe state of system is expressed by assigning definite values to the thermodynamic properties of the system. When one or more of these variables are changed, the system also undergoes a change. The method of operation which can bring about a change in the system is called thermodynamic process. Some examples of a thermodynamic process are heating, cooling, expansion, compression, fusion, vaporization etc. The processes can be classified depending on how the processes are carried out in different ways and under different conditions.Reversible process: The process in which the system and surrounding can be restored to the initial state from the final state without producing any changes in the thermodynamic properties of the universe is called a reversible process. There are two important conditions for the reversible process to occur. Firstly, the process should occur infinitesimally slowly and secondly throughout the process, the system and surroundings must be in equilibrium with each other.Irreversible Process: The process in which the system and surrounding cannot be restored to the initial state from the final state is called an irreversible process. All the processes occurring in nature are irreversible processes. During the irreversible process the system and surroundings are not in equilibrium with each other.Adiabatic process: If the change takes place in such a way that there is no exchange of heat (q) between the system and surrounding during the process, it is called adiabatic process. This condition is attained by thermally insulating the system. In an adiabatic process if work is done by the system its temperature decreases, if work is done on the system its temperature increases, because, the system cannot exchange heat with its surroundings. For an adiabatic process q = 0Isothermal process: If the change takes place in such a way that the temperature remains constant throughout from initial to final state, it is called an isothermal process. The system exchanges heat with its surrounding and the temperature of the system remains constant. For this purpose the experiment is often performed in a thermostat. For an isothermal process ΔT = 0Isobaric process: In this process, the pressure of the system remains constant during its change from the initial to final state. For an isobaric process ΔP= 0.Isochoric process: In this process, the volume of system remains constant during its change from initial to final state. Combustion of a fuel in a bomb calorimeter is an example of an isochoric process. For an isochoric process, ΔV= 0.Cyclic process: If as a result of series of changes, a system returns to its initial state, its thermodynamic parameters would also return to their original values. This process is known as a cyclic process. For a cyclic process ΔU = 0, ΔH = 0, ΔP = 0, ΔV = 0, ΔT = 06.5 State function and Path functionState function: A state function is a thermodynamic property of a system, which has a specific value for a given state and does not depend on the path (or manner) by which the particular state is reached. Example: pressure (P), volume (V), temperature (T), internal energy (U), enthalpy (H), etc.Path functions: A path function is a thermodynamic property of the system whose value depends on the path by which the system changes from its initial to final states. Example: Work (w), Heat (q), etc. Work (w) will have different values if the process is carried out reversibly or irreversibly.6.6 Internal Energy (U)The internal energy of a system is equal to the energy possessed by all its constituents (atoms, ions and molecules, etc.). It may be chemical, electrical, mechanical or any other type of energy, the sum of all these is called internal energy of the system. In thermodynamics, one is concerned only with the change in internal energy (ΔU) rather than the absolute value of energy. It may change, when heat passes into or out of the system, work is done on or by the system, matter enters or leaves the system. 6.6.1 Features of internal energy (U) The internal energy of a system is an extensive property. Being a state function, it depends only upon the state variables (T, P, V, n) of the system. The change in internal energy does not depend on the path by which the final state is reached. The change in internal energy of a system is expressed as ∆U= (Uf – Ui), where Uf and Ui are the internal energy of the system in the final state and initial states respectively. In a cyclic process, there is no internal energy change. ∆U(cyclic) = 0 If (Uf < Ui), then ΔU would be negative and if (Uf > Ui), then ΔU would be positive. 6.7 Heat (q)The heat (q) is regarded as energy in transit across the boundary separating a system from its surrounding. So, heat is flow of energy from one system to another on account of difference in temperature between them. It is a path function. The SI unit of heat is joule (J) which is defined as the work done by a force of one Newton through a displacement of one meter (J= N.m). Heat quantities are generally measured in calories (cal). A calorie is defined as the quantity of heat required to raise the temperature of 1 gram of water by 1 oC at a pressure of one standard atmospheric pressure.Sign convention of heat (IUPAC): If heat flows into the system from the surrounding, internal energy of a system increases. Hence, it is taken to be positive (+q). If heat flows out of the system into the surrounding, internal energy of the system decreases. Hence, it is taken to be negative (–q).6.8 Work (w)Work is defined as the force (F) multiplied by the displacement (Δx).−w = F. ΔxIf a system loses energy by working against external mechanical force, it is said to do work. The negative sign is introduced to indicate that the work has been done by the system by spending a part of its internal energy. Work is a path function. The SI unit of work is joule (J). We often use kilojoule (kJ) for large quantities of work. 1 kJ = 1000 J. Sign convention of work (IUPAC): If work is done by the system, the energy of the system decreases, hence by convention, work is taken to be negative (−w). If work is done on the system, the energy of the system increases, hence by convention, the work is taken to be positive (+w).Note: Work is transfer of energy without a temperature difference while heat is transfer of energy due to temperature difference only. Heat should be carefully distinguished from work.6.9 Pressure - Volume work (mechanical work)Chemical reactions may involve the generation of gases capable of doing mechanical work or the generation of heat. It is important to quantify these changes and relate them to the changes in the internal energy. In elementary thermodynamics the only type of work generally considered is the work done in expansion (or compression) of a gas, known as pressure-volume work (PV work).6.9.1 Work involved in expansion and compression processes
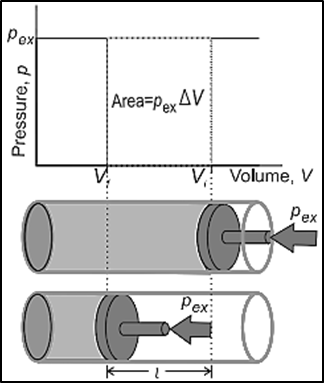
In most thermodynamic calculations, we deal with the evaluation of work involved in the expansion or compression of gases. The essential condition for expansion or compression of a system is that there should be difference between external pressure (Pext) and internal pressure (Pint).For understanding pressure-volume work, let us consider a cylinder which contains ’n’ moles of an ideal gas fitted with a frictionless piston of cross sectional area A. The total volume of the gas inside is Vi and pressure of the gas inside is Pint. If the external pressure Pext is greater than Pint, the piston moves inward till the pressure inside becomes equal to Pext. Let this change be achieved in a single step and the final volume be Vf. In this case, the work is done on the system (+w). It can be calculated as followsw = –F.l …………… (1) as work = force x distanceHere, l is the distance moved by the piston during the compression and F is the force acting on the gas.F= Pext . A ………… (2) as force = pressure x areaHere, A = Cross-sectional area of the pistonSubstituting equation (2) in equation (1)w = – (Pext . A). l = – Pext . (Vf - Vi) as (A.l) = (area x distance moved) = change in volume = (Vf – Vi)w = – Pext . (–∆V) = Pext .∆V as Vf < ViSince work is done on the system, it is a positive quantity.So, we can concludeFor expansion, Vf > Vi so that ( Vf – Vi ) is positive and hence w is negative.For compressions, Vf < Vi so that (Vf –Vi) is negative and hence w is positive.It is the external pressure against gas expands i.e. Pext and not the pressure of the gas that is used in evaluating the pressure volume work done. The final equation shows that work W depends only on the change in volume (ΔV) and opposing pressure (Pext) and not on the quantity of the gas (i.e. it is independent of the number of moles) and temperature of the gas (T). The work done by the system in a process depends on the way or manner in which it is carried out. Work, therefore, is not a state function. It is a path function.
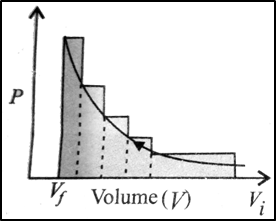
If the pressure is not constant at every stage of compression, but changes in number of finite steps, work done on the gas will be summed over all the steps and will be equal to − ∑p.V as shown in the figure. Work done on the gas is represented by the shaded area.
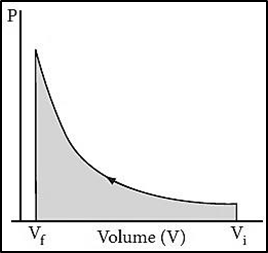
If the pressure is not constant, but changes during the process is such that it is always infinitesimally greater than the pressure of the gas, then, at each stage of compression, the volume decreases by an infinitesimal amount, dV. Here, changes are occurring in infinitesimally small steps (reversible conditions) during compression from Vi to Vf, the P-V plot looks like the figure. Work done on the gas is represented by the shaded area. Such processes are called reversible processes. In such a case we can calculate the work done on the gas by the relationWrev = -∫vfvi Pext dV ......................(3)Processes other than reversible processes are known as irreversible processes.6.9.2 Work done in isothermal reversible expansion of an ideal gasIn a compression process, Pext is always greater than the pressure of the system, i.e Pext = (Pint + dP). In an expansion process,Pext is always less than the pressure of the system i.e. Pext = (Pint - dP). In general case we can write, Pext = (Pint ± dP).If we relate the work term to the internal pressure of the system, we can relate work to internal pressure of the system under reversible conditions by writing equation (3) as follows: Wrev = - ∫vfvi Pext dV" = - ∫vfvi (Pint ± dp)Since (dp.dv) is very small, the equation (3) can be written as Wrev = - ∫vfviPint dV ……………….(4)For a given system with an ideal gas, Pint .V = nRT, or Pint = nRT/VHere, n = number of moles of the gas, R = universal gas constant, T = temperature in kelvinSubstituting the value of Pint in equation (4), we getWrev = - ∫vfvinRT
vdVWrev = - nRT∫vfvidv
vWrev = - nRT ln (vf
vi)Wrev = - 2.303nRT log (vf
vi)……………..(5)If Vf >Vi (expansion), the sign of work done by the process is negative. If Vf <Vi (compression) the sign of work done on the process is positive. Work done in an isothermal reversible expansion is maximum work.Since vf
vi=pi
Pf from Boyle’s lawEquation (5) can also be written as Wrev = - 2.303nRT log (pi
pf)In case of free expansion of an ideal gas that is expansion of a gas in vacuum (Pex = 0), no work is done whether the process is reversible or irreversible6.10 First Law of ThermodynamicIf we consider the general case in which a change of state is brought about both by work (w) being done by/on the system and by transfer of heat (q, absorption or evolution), then change in internal energy for this case is∆U = q + w …………(6)This is the mathematical statement of the First Law which states that “the total energy of an isolated system remains constant though it may change from one form to another”. For a given change in state, q and w can vary depending on how the change is carried out. However, ∆U will depend only on initial and final state and independent of the way the change is carried out. If there is no transfer of energy as heat or as work (isolated system) i.e., if w = 0 and q = 0, then ∆U = 0.The first law of thermodynamics can also be stated in a different way such as Whenever energy of a particular type disappears, an equivalent amount of another type must be produced. The total energy of a system and surrounding remains constant (or conserved) Energy can neither be created nor destroyed, but may be converted from one form to another. The change in the internal energy of a closed system is equal to the energy that passes through its boundary as heat or work. Heat and work are two ways of changing a system’s internal energy.6.10.1 Different cases of the mathematical statement of the first lawCase 1: For a isothermal expansion of an ideal gas, ∆U = 0. Equation (6) becomes ∆U = q + w∴ q = -wIn other words, during an isothermal process, the amount of heat absorbed by the system is equal to work done by the system.For isothermal irreversible changeq = -w = pext (Vf – Vi) For isothermal reversible changeq=Wrev = - 2.303nRT log (vf
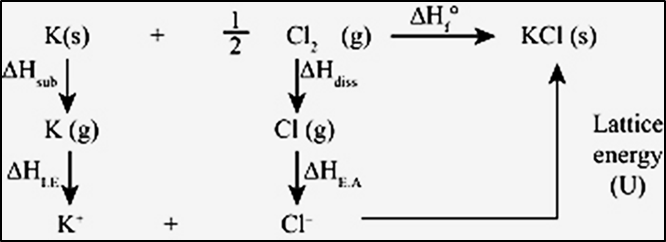
vi)Case 2: For an isochoric process (no change in volume) there is no work of expansion. i.e. ΔV = 0. Equation (6) becomesΔU = q + w = q + PΔVΔU = qvIn other words, during an isochoric process, the amount of heat supplied to the system is converted to its internal energy.Case 3: For an adiabatic process there is no change in heat, i.e. q = 0. Hence, q = 0. Equation (6) becomes ∆U = q + wΔU = wadIn other words, in an adiabatic process, the decrease in internal energy is exactly equal to the work done by the system on its surroundings.Case 4: For an isobaric process, there is no change in the pressure. P remains constant. Hence, equation (6) becomes ∆U = q + w∆U = q - P∆V……….(7)In other words, in an isobaric process a part of heat absorbed by the system is used for PV expansion work (work done by the system) and the remaining is added to the internal energy of the system.6.11 Enthalpy (H)The heat absorbed at constant volume (isochoric process) is equal to change in the internal energy i.e., ∆U = qv. But most of chemical reactions are carried out not at constant volume, but under constant atmospheric pressure. Enthalpy is such a state function which may be suitable under these conditions. At constant pressure, we can write equation (7) as ∆U = qp – P∆V where qp is heat absorbed by the system and –P∆V represent expansion work done by the system. Suppose the initial state is denoted by subscript 1 and final state by 2. We can rewrite the above equation asU2–U1 = qp– P(V2 – V1)On rearranging, we getqp = (U2+ PV2) – (U1 + PV1) …………(8)Enthalpy is the sum of the internal energy (U) of a system and the product of pressure and volume of the system. H = U + PV……………(9)Finally the equation (8) becomesqp = H2 - H1q is a path dependent function but H is a state function as it depends on U, P and V, all of which are state functions. Therefore, ∆H is independent of path. Hence, qp is also independent of path. When a process occurs at constant pressure, the heat involved (either released or absorbed) is equal to the change in enthalpy.For finite changes at constant pressure, equation (9) can be written as ∆H = ∆U + ∆PVSince P is constant, we can write∆H = ∆U + P∆V ……………(10)It reflects the capacity to do mechanical work and the capacity to release heat by the system. In an endothermic reaction heat is absorbed by the system from the surroundings that is q > 0 (positive). Therefore, ΔH is also positive. In an exothermic reaction heat is evolved by the system to the surroundings that is, q < 0 (negative). If q is negative, then ΔH will also be negative. At constant volume (∆V = 0), ∆U = qv, therefore equation (10) becomes∆H = ∆U = qvThe difference between ∆H and ∆U is not usually significant for systems consisting of only solids and/or liquids as solids and liquids do not suffer any significant volume changes upon heating. The difference is significant when gases are involved. Let us consider a reaction involving gases. Consider a closed system of gases which are chemically reacting to form gaseous products at constant temperature and pressure with Vi and Vf as the total volumes of the reactant and product gases respectively, and ni and nf as the number of moles of gaseous reactants and products, then,For reactants (initial state) : PVi = ni RT For products (final state) : PVf = nf RT P(Vf -Vi) = (nf - ni) RTP∆V= ∆ng RT Substituting this value of P∆V in equation (10)∆H = ∆U + ∆ngRT6.11.1 Enthalpy Changes for different types of reactions and Phase transitionsThe heat or enthalpy changes accompanying chemical reactions are expressed in different ways depending on the nature of the reaction.Standard heat of formationIt is the change in enthalpy that takes place when one mole of a compound is formed from its elements, present in their standard states (298 K and 1 bar pressure). By convention the standard heat of formation of all elements is assigned a value of zero.Fe (s) + S (s) → FeS (s) ΔHof = - 100.42 kJ mol-12C (s) + H2 (g) → C2H2 (g) ΔHof = + 222.33 kJ mol-1Standard enthalpy of reactionIt is the change in enthalpy for a reaction when all the reactants and products are in their standard states (298 K and 1 bar pressure). We can calculate the enthalpy of a reaction under standard conditions from the values of standard enthalpies of formation of various reactants and products. The standard enthalpy of reaction is equal to the difference between standard enthalpy of formation of products and the standard enthalpies of formation of reactants.ΔHor = ΣΔHof(products) - ΣΔHof (reactants)For a general reaction like aA + bB → cC +dDΔHor = {c.ΔHof(C) + d.ΔHof(D)} - {a.ΔHof(A) + b.ΔHof(B)}Enthalpy Changes during Phase transitionPhase transition such as melting, vaporization, sublimation occurs at constant temperature and pressure and they involve enthalpy change.Standard enthalpy of VaporizationThe enthalpy change involved in the vaporization of one mole of a liquid at constant temperature and under standard pressure (1bar) is called its standard enthalpy of vaporization or molar enthalpy of vaporization, ∆HovapExample – the standard enthalpy of vaporization of water is shown below.H2O (l) → H2O (g); ∆Hovap = +40.79 kJ mo-1Standard enthalpy of FusionThe enthalpy change involved in the melting of one mole of a solid substance in standard state is called standard enthalpy of fusion or molar enthalpy of fusion, ∆Hofus. Melting of a solid is endothermic, so all enthalpies of fusion are positive. Example – the standard enthalpy of fusion of ice is shown below.H2O (s) → H2O (l); ∆Hofus = 6.00 kJ moI-1Standard enthalpy of CombustionIt is the enthalpy change involved when one mole of a compound is completely burnt in oxygen under standard conditions (298 K and 1 bar pressure). Combustion reactions are exothermic in nature. Example – the standard enthalpy of combustion of ethane (C2H6) is shown below.C2H6 (g) + 7/2 O2 (g) → 2CO2 (g) + 3H2O (l) ∆Hocom = -1558.3 kJ moI-1Standard enthalpy of atomizationIt is the amount of enthalpy change when one mole of a substance (molecule, element or compound) is dissociated into its atoms under standard conditions. Enthalpy of atomization is always a positive value. Example - the standard enthalpy of atomisation of Na, F2 and CH4 are shown below.Na (s) → Na (g) ∆Hoa = 108.4 kJ mol-1F2 (g) → 2F (g) ∆Hoa = 79 kJ mol-1CH4 (g) → C (g) + 4H (g) ∆Hoa = 1665 kJ mol-1For diatomic molecules like F2, the enthalpy of atomization is equal to the enthalpy of bond dissociation. For sodium (Na) metal, the enthalpy of atomization is the sum of the enthalpy of fusion and the enthalpy of vaporization of sodium. For any elemental solid, the enthalpy of atomization is the same as the enthalpy of sublimation.Bond enthalpyEnergy is required to break a chemical bond and energies are released when a bond is broken. Two different terms are used in thermodynamics when enthalpy changes associated with chemical bonds are considered: (i) Bond dissociation enthalpy and (ii) Mean bond enthalpy Diatomic moleculesThe bond dissociation enthalpy is the change in enthalpy when one mole of particular covalent bonds is broken in the gas phase. It is the same as the enthalpy of atomization of diatomic chlorine and true for all diatomic molecules. Cl2 (g) → 2Cl (g); ∆Hocl-cl = 242.7 kJ mol-1Polyatomic moleculesIn polyatomic molecules, the term mean or average bond enthalpy is used as bond dissociation enthalpy is different for different bonds within the same molecule. It is calculated by dividing the total bond dissociation enthalpy by the number of bonds broken. For example, the overall enthalpy change for the following equation is shown. CH4 (g) → C (g) + 4H (g) ∆Ho = 1665 kJ mol-1Though all the four C-H bonds are identical in bond length and energy, the energies required to break the individual C-H bonds in each successive step are different.CH4 (g) → CH3 (g) + H (g); ∆HoC-H = +427 kJ mol-1CH3 (g) → CH2 (g) + H (g); ∆HoC-H = +439 kJ mol-1CH2 (g) → CH (g) + H(g); ∆HoC-H = +452 kJ mol-1CH (g) → C (g) + H (g); ∆HoC-H = +347 kJ mol-1In this case mean bond enthalpy will be used which is (¼ x 1665) = 416.0 kJ mol-1. The mean C–H bond enthalpies may differ slightly from compound to compound where C-H bond is present but the difference is not significant.For any chemical reaction, the estimated change in enthalpy is the sum of the bond enthalpies of the bonds broken (bonds present in reactants) minus the sum of the bond enthalpies of the bonds formed (bonds present in products).∆Hor = ∑bond enthalpies (reactants) - ∑bond enthalpies (products)This relationship is useful when the required values of enthalpy of formations (∆Hof) are not available.Lattice enthalpyIt is the enthalpy change involved to completely separate one mole of the solid ionic compound into constituent gaseous ions. It is a measure of the strength of an ionic compound. The greater the lattice enthalpy, the stronger the forces present between ions. It can also be called Lattice dissociation enthalpy.KCl (s) → K+ (g) + Cl- (g) ∆Holattice= +716.8 kJ mol-1Lattice formation enthalpy is the amount of energy released when a lattice is formed from its scattered gaseous ions. For KCl, the value is -716.8 kJ mol-1. The numerical value is same as that of Lattice dissociation enthalpy.As Lattice enthalpy cannot be measured directly, we need to take the help of the Born-Haber cycle based on Hess’s Law. Hess’s Law The law states that “the change in enthalpy for a reaction is the same whether the reaction takes place in one or a series of steps”. This law is a manifestation that enthalpy is a state function.Let us suppose substance ‘A’ is converted into D directly.A → D ΔH = Q kJSuppose the same change is brought about in different steps,i) A → B ΔH1 = Q1 kJii) B → C ΔH2 = Q2 kJiii) C → D ΔH3 = Q3 kJNow, according to Hess’s law,ΔH = ΔH1 + ΔH2 + ΔH3Q = Q1 + Q2 + Q3Thus, Hess’s law implies that change in enthalpy of a chemical reaction depends upon the initial and final state of a chemical reaction irrespective of the number of steps involved in a chemical reaction.Calculation of Lattice enthalpyWe can calculate the lattice enthalpy of KCl (s) by following the steps given below;Sublimation energy of K metal K (s) → K (g) ΔHsub = 89 kJ/molIonization energy of K atomK (g) → K+ (g) + e ΔHIE = 418.8 kJ/molBond dissociation energy of diatomic Cl2½ Cl2 → Cl (g) ½ ΔHdis = (½ x 242.7 kJ/mol) = 121.4 kJ/molElectron gain by Cl atom Cl (g) + e → Cl- (g) ΔHEA = Electron affinity = -349 kJ/molEnthalpy of formation of KCl (s) Na (s) + ½ Cl2 (g) → NaCl (s) ΔHof = -436.6 kJ/mol U = Lattice formation enthalpy Applying Hess’s Law, we get ΔHof = ΔHsub + ΔHdis + ΔHIE + ΔHEA + U
Born-Haber cycle of KClPutting all the values in the above equation we can get the value of U which is -716.8 kJ/mol. There will be a negative sign as we have considered the enthalpy change when 1 mole of solid KCl is formed from its separated gaseous ions (Lattice formation enthalpy).Standard enthalpy of solutionIt is the enthalpy change ∆Hosol when one mole of a substance is dissolved in a specified amount of solvent at constant pressure under standard conditions. The enthalpy of solution at infinite dilution is the enthalpy change on dissolving the substance in an infinite amount of solvent when the interactions between the ions or solute molecules are negligible.
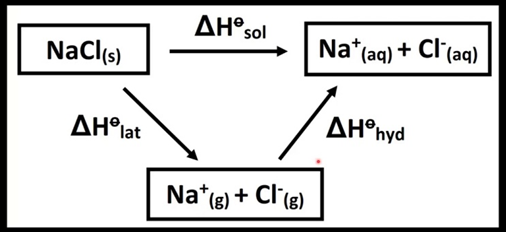
We can use the value of lattice enthalpy to calculate enthalpy of solution from the expression. ∆Hosol =∆Holattice+∆HohydHere ∆Hohyd is the enthalpy of hydration which is the enthalpy change when one mole of ions undergoes hydration.Standard enthalpy of DilutionIt is the enthalpy change involved when a solution containing one mole of solute is diluted from higher concentration to lower concentration under standard condition. Suppose, the enthalpy change for dissolving one mole of gaseous HCl in 25 and 40 moles of water (symbol aq. used for water) are represented asHCl (g) + 25 aq. → HCl.25 aq. ∆H = –72.03 kJ / molHCl (g) + 40 aq. → HCl.40 aq. ∆H = –72.79 kJ / molSubtracting the first equation from the second equation we getHCl.25 aq. + 15 aq. → HCl.40 aq. and ∆H = [ –72.79 – (–72.03)] kJ/mol = – 0.76 kJ/molThis value (–0.76 kJ/mol) is enthalpy of dilution (of ∆Hodil). The enthalpy of dilution of a solution is dependent on the original concentration of the solution and the amount of solvent added.Difference between Dissolution and DilutionThough dissolution and dilution processes are closely related, it is important to note that they are not the same. Despite the fact that both processes have similar final stages, they differ in their initial stages. Dissolution is the process of dissolving a solute into a solvent to make a solution. Dilution is the process of decreasing the concentration of a solute by mixing it with more solvent. If the initial state of the component is a pure liquid (presuming the solution is liquid), the dilution process is equal to its dissolution process and the enthalpy of dilution is the same as the enthalpy of solution. This is because the dilution process in this scenario is the same as the dissolution process for the component being dissolved.Standard enthalpy of NeutralizationIt is the change in enthalpy when solutions of an acid and an alkali react together under standard conditions to produce salt and 1 mole of water. It is always measured per mole of water formed. The enthalpy change for a neutralization reaction is always negative, indicating that heat is always liberated. It is a special case of the enthalpy of reaction.HCl (aq.) + NaOH (aq.) NaCl (aq.) + H2O (l)It has been experimentally observed that the heat of neutralization for the above reaction is – 57.9 kJ/mol. Neutralizations between all the strong acids and strong bases show this value of enthalpy. This constancy of heat of neutralization of a strong acid and strong base can be explained on the basis of ionic theory.According to the theory of electrolytes all strong acids and strong bases are completely ionized in solution. The above neutralization reactions in aqueous solutions can be written in the ionic from as:Na+ (aq) + HO- (aq) + H+ (aq) + Cl- (aq) → Na+ (aq) + Cl- (aq) + H2O (l)The reaction between any strong acid and a strong base directly involves H+ ions combining with HO- ions. The cation from the base (Na+) and the anion from the acid (Cl-) are spectator ions. Cancelling the spectator ions from both sides, we get the net ionic equation asH+ (aq) + HO- (aq) → H2O (l)Here, one mole of H+ (aq) ions react with one mole of HO- (aq) ions to form one mol of H2O (l). Hence, the heat of neutralization is always same, i.e., -57.9 kJ mol-1 of water formed. If however, either the acid or the base or both are weak, the heat of neutralization will not be equal to -57.9 kJ mol-1 as weak electrolytes do not dissociate completely.Calorific value of food and fuelsThe calorific value is defined as the amount of heat produced in calories (or joules) when one gram of the substance is completely burnt. The calorific value of food indicates the total amount of energy, a human body could generate during its metabolism which is expressed in Kcal/g. When the calorific value of a fuel is high, more amount of heat energy is produced, and hence, more is its efficiency.
Fuel
Calorific value (Kcal/kg)
Food category
Approx.Calorific value (Kcal/g)
Coal
5377
Dietary fibre
2
Kerosene
10277
Polyhydric alcohols
2.4
Methane
13289
Alcohols
7
Methanol
4732
Carbohydrates
4
Wood
4780
Protein
4
Hydrogen
34178
Fats
9
Calorific Value of some food and fuelsImportance of Calorific ValueIt is very important to have knowledge of the calorific value of fuel to carry out our day-to-day activities. The gas shippers and suppliers require this information to bill gas consumers. It also helps to determine transportation charges of gas shippers and suppliers. The human body requires energy to carry out daily activities. Without this, the body would stop working and the cells in the body would die. A certain amount of calories daily is ideal to lead a healthy life. Too high or too low calorie consumption eventually leads to health problems.6.12 Thermochemical EquationsA thermochemical equation is a balanced stoichiometric chemical equation that includes the enthalpy change (ΔH). The following conventions are followed in thermochemical equations: The coefficients in a balanced thermochemical equation refer to number of moles of reactants and products involved in the reaction. The enthalpy change of the reaction ΔHr has to be specified with appropriate sign and unit. When the chemical reaction is reversed, the value of ΔH is reversed in sign with the same magnitude. The physical states (gas, liquid, aqueous, solid in brackets) of all species are important and must be specified in a thermochemical reaction, since ΔH depends on the physical state of reactants and products. If the thermochemical equation is multiplied throughout by a number, the enthalpy change is also multiplied by the same number as enthalpy is an extensive property. The negative sign of ΔHr indicates that the reaction is exothermic and the positive sign of ΔHr indicates an endothermic reaction. Unless mentioned, ΔH values are for the standard state of the reactants and products.6.13 Heat capacitiesWhen heat (q) is supplied to a system, the molecules in the system absorb the heat and hence their kinetic energy increases, which in turn raises the temperature of the system from T1 to T2. This increase in temperature (ΔT) is directly proportional to the amount of heat absorbed and the mass of the substance. q ∝ ΔTq = C.ΔT or C = q/ΔTThis C is called heat capacity defined as the amount of heat energy required to raise the temperature of a given quantity of matter by one degree (Celsius or Kelvin). Heat capacity for a given matter depends on its quantity and hence it is an extensive property. The unit of heat capacity is J/K or J/ºC. When C is large, a given amount of heat results in only a small temperature rise and in that case a lot of energy is needed to raise its temperature. Specific heat capacityScientists needed a quantity that has no dependence on the quantity of matter under consideration for thermodynamic studies. This made them define specific heat capacity which is an intensive property as it is independent of the size of the matter. When m = 1 unit mass and T = 1 K or 1 ºC, then c = q and the heat capacity is referred as specific heatcapacity (c). So, specific heat capacity is “the amount of heat energy required to raise the temperature of a substance per unit of mass”. The SI unit of specific heat capacity is JK-1kg-1. Molar heat capacityThe heat capacity for 1 mole of substance is called molar heat capacity (Cm = C/n, n = number of moles) which is defined as “the amount of heat absorbed by one mole of the substance to raise its temperature by 1 K or 1 ºC”. The SI unit of molar heat capacity is JK-1mol-1. Molar heat capacity is very similar to specific heat capacity but measures per mole instead of per gram of substance. Molar heat capacity is an intensive property and hence it doesn’t vary with the amount of substance. Relationship between Cp and Cv for an Ideal gasThe molar heat capacities can be expressed either at constant volume (Cv) or at constant pressure (Cp). Molar heat capacity at constant volume can be expressed as qV = ∆U = Cv∆T Molar heat capacity at constant pressure can be expressed as qp = ∆H = Cp∆T. Similarly for n moles of an ideal gas we get ∆U = nCv∆T and ∆H= nCp∆T.From the definition of enthalpy ∆H = ∆U + ∆(PV) For 1 mole of an ideal gas PV = RT So, we can write ∆H = ∆U + ∆RT = ∆U + R∆TPutting the values of ∆H and ∆U in the above equationCp∆T = Cv∆T + R∆T or Cp = Cv + Ror Cp - Cv = RAt constant pressure processes, a system has to do work against the surroundings (P∆V≠0). Hence, the system would require more heat to effect a given temperature rise than at constant volume. Hence, Cp is always greater than Cv6.14 Kirchhoff Equation: Influence of temperature on ∆H and ∆UThe change in enthalpy and change in internal energy of any physical or chemical process varies with temperature at constant pressure. If a system undergoes change from state 1 to state 2, then Kirchhoff’s equations are∆H2 - ∆H1 = ∆Cp(T2-T1) ∆U2 - ∆U1 = ∆Cv(T2-T1) (assuming heat capacities are not dependent on temperature)∆H1 and ∆H2 are the enthalpy change for the process at temperature T1 and T2 respectively. ∆U1 and ∆U2 are the internal energy change for the process at temperature T1 and T2 respectively. ∆Cp = difference of heat capacities of products and reactions at constant pressure∆Cv = difference of heat capacities of products and reactions at constant volume6.15 Necessity of Second Law of Thermodynamics
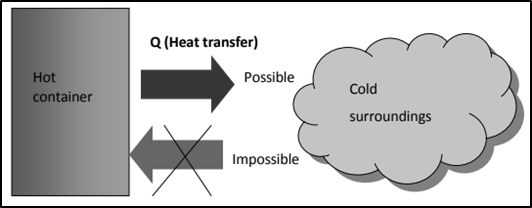
The first law places no restriction on the direction of a process, and satisfying the first law does not guarantee that the process will occur. Thus, we need another general principle (second law) to identify whether a process can occur or not. Heat transfer from a hot container to the cold surroundings is possible but the reveres process (although satisfying the first law) by absorbing heat from surrounding on its own does not occur spontaneously. If the heat energy is supplied to cold water, then only it will become hot. i.e. the change that does not occur spontaneously and can be driven by supplying energy only.First law also tells that there is an exact equivalence between various forms of energy and that heat gained is equal to heat loss. Practically it is not possible to convert the heat energy into an equivalent amount of work. To explain this, another law is needed (second law of thermodynamics). The second law of thermodynamics helps us to predict whether the reaction is feasible or not and also tell the direction of the flow of heat. It also tells that energy cannot be completely converted into equivalent work. A process can occur when and only when it satisfies both the first and the second laws of thermodynamics.SpontaneitySpontaneity means ‘having the potential to proceed without the assistance of external agency’. However, it does not tell about the rate of the reaction or process. Another aspect of spontaneous reaction or process, as we see is that these cannot reverse their direction on their own. A spontaneous process is an irreversible process and may only be reversed by some external agency.6.15.1 EntropyThe second law of thermodynamics introduces another state function called entropy. Entropy is a measure of the molecular disorder (randomness) of a system. But thermodynamic definition of entropy is concerned with the change in entropy that occurs as a result of a process. It is defined as, ΔS = qrev
TFor any spontaneous irreversible process ΔS = qirrev
TThe entropy (S) is equal to heat energy exchanged (q) divided by the temperature (T) at which the exchange takes place. Therefore, The SI unit of entropy is JK-1.The spontaneity of reaction can be determined by the following relations. If ΔSTotal > 0, the process is spontaneous If ΔSTotal < 0, the process is non-spontaneous If ΔSTotal = 0, the process is in equilibrium6.15.2 Entropy statement of the second law of thermodynamicsFor an isolated system the change in energy remains constant and increase in entropy in such systems is the natural direction of a spontaneous change. The second law of thermodynamics can be expressed in terms of entropy. i.e “the entropy of an isolated system increases during a spontaneous process”.Mathematically, the second law of thermodynamics is represented as for an irreversible process such as spontaneous expansion of a gas,∆Stoal > 0 or ΔSuniversity> 0 [ΔSuniversity = the change in the entropy of the universe]∆Stotall > ∆Ssystem + ∆Ssurroundingi.e. ∆Suniverse > (∆SSystem+ ∆Ssurrounding)Entropy is a measure of the randomness of the system, or it is the measure of energy or chaos within an isolated system. It can be considered a quantitative index that describes the quality of energy. For a reversible process such as melting of ice,∆Ssystem = - ∆Ssurrounding∆Suniverse = 06.16 Gibbs free energyOne of the important applications of the second law of thermodynamics is to predict the spontaneity of a reaction under a specific set of conditions. In our daily life, we observe many spontaneous physical and chemical processes which cannot take place in opposite direction spontaneously, such as1. A waterfall runs downhill, but never uphill, spontaneously.2. A lump of sugar dissolves spontaneously in a cup of coffee, but never reappears in its original form spontaneously.3. Heat flows from hotter object to a colder one, but never flows from colder to hotter object spontaneously.4. The expansion of a gas into an evacuated bulb is a spontaneous process; the reverse process that is gathering of all molecules into one bulb is not. A large number of exothermic reactions are spontaneous such as combustion of methane, acid-base neutralization reaction. However, some endothermic processes are also spontaneous. For example ammonium nitrate dissolves in water spontaneously though this dissolution is endothermic. For an isolated system, it is the total entropy change, ∆Stotal which decides the spontaneity of the process. But most of the chemical reactions fall into the category of either closed systems or open systems and there are changes in both enthalpy and entropy. Exothermicity favors the spontaneity but does not guarantee it. Neither decrease in enthalpy nor increase in entropy alone can determine the direction of spontaneous change for these systems. For this purpose, we define a new thermodynamic function the Gibbs energy or Gibbs function (G) which finds useful in predicting the spontaneity of a reaction. This function was developed by Josiah Willard Gibbs who originally termed this energy as the “available energy” to do work in a system. Gibbs free energy is defined as belowG = H - TS Gibbs free energy (G) is an extensive property and it is a single valued state function. Let us consider a system which undergoes a change of state from state (1) to state (2) at constant temperature.G2 – G1 = (H2 – H1) – T(S2 – S1)ΔG = ΔH – T ΔSNow let us see now, how ΔG is related to reaction spontaneity. We know thatΔStotal = ΔSsys + ΔSsurrFor a reversible process (equilibrium), the change in entropy of universe is zero (ΔStotal = 0, ∴ ΔSsys = −ΔSsurr). Similarly, for an equilibrium process ΔG=0If the system is in thermal equilibrium with the surrounding, then the temperature of the surrounding is same as that of the system. Also, increase in enthalpy of the surrounding is equal to decrease in the enthalpy of the system.ΔHsurr = - ΔHsysFor a spontaneous process; ΔStotal > 0. So, we can writeΔSsystem + ΔSsurrounding > 0ΔSsys + ∆Hsurr
T > 0ΔSsys - ∆Hsys
T > 0TΔSsys - ΔHsys > 0-(ΔHsys - TΔSsys) > 0-ΔGsys > 0Usually the subscript ‘system’ is dropped and we simply write this equation as-ΔG > 0ΔG < 0ΔG = (ΔH – TΔS) < 0The spontaneity of reaction can be determined by following relations. If ΔG < 0, the process is spontaneous If ΔG > 0, the process is non-spontaneous If ΔG = 0, the process is in equilibriumsΔHsys is the enthalpy change of a reaction; TΔSsys is the energy which is not available to do useful work. So, ΔG is the net energy available to do useful work and is thus a measure of the ‘free energy’. For this reason, it is also known as the free energy of the reaction. 6.16.1 Gibbs free energy and the net work done by the systemFor any system at constant pressure and temperatureΔG = ΔH – TΔSΔG = (ΔU + PΔV) – TΔS ……….(11) as ΔH = (ΔU + PΔV)Now, from first law of thermodynamics, we getΔU = q + wFrom second law of thermodynamics, we getΔS = q/TSo, putting the values of ΔU and ΔS in equation (11) of ΔG,ΔG = (q + w + PΔV) – T(q/T) ΔG = w + PΔVor −ΔG = (−w) − PΔV (work done by the system = -w)Where PΔV is the mechanical work done by the system (expansion against a constant external pressure) and w is the maximum total work. The decrease in free energy (–ΔG) accompanying a process taking place at constant temperature and pressure is equal to the maximum work obtainable from the system other than the work of expansion or non PV work. 6.16.2 Effect of temperature on the spontaneity of a processIt is assumed here that ΔH and ΔS will remain the way indicated for all temperatures though it is not necessary. The spontaneity of a chemical reaction is only the potential for the reaction to proceed as written. However, the rate of such processes is determined by kinetic factors also outside of thermodynamic prediction.
ΔH
ΔS
ΔG = ΔH - TΔS
Spontaneity
-
+
-
Spontaneous at all temperatures
+
-
+
Non-spontaneous at all temperatures
-
-
- (low T)
Spontaneous at low temperature
+ (high T)
Non-spontaneous at high temperature
+
+
- (high T)
Spontaneous at high temperature
+ (low T)
Non-spontaneous at low temperature
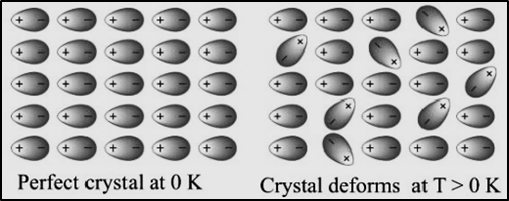
6.16.3 Free energy change (ΔG) and equilibrium constant (Keq)In a reversible process, the system is in perfect equilibrium with its surroundings at all times. A reversible chemical reaction can proceed in either direction simultaneously, so that a dynamic equilibrium is set up. This means that the reactions in both the directions should proceed with decrease in free energy, which is impossible. It is possible only if at equilibrium, the free energy of a system is minimum. Let us consider a general equilibrium reactionaA + bB ⇌ cC + dDThe free energy change of the above reaction in any state (ΔG) is related to the standard free energy change of the reaction (ΔGº) asΔG = ΔGº + RTlnQWhere ΔG° indicates that all reactants and products are in their standard states, Q is reaction quotient and is defined as the ratio of concentration of the products to the concentrations of the reactants under non-equilibrium condition. When equilibrium is attained, there is no further free energy change i.e. ΔG = 0 and Q becomes equal to equilibrium constant Keq. Hence the above equation becomes.ΔGº = –RTlnKeq = -2.303RTlogKeqWe also know thatΔGº = ΔHº – TΔSº = − RTlnKeqWith the above equation, it is possible to obtain an estimate of ∆Gº from the measurement of ∆Hº and ∆Sº and then calculate Keq at any temperature. If K is measured directly in the laboratory, value of ∆Gº at any other temperature can be calculated.6.17 Helmholtz energyIf a system is isothermal and closed, with constant pressure, it is describable by the Gibbs energy (G) whereas if a system is isothermal and closed with constant volume, it is describable by the Helmholtz energy (A). Though IUPAC (the International Union of Pure and Applied Chemistry) officially refers to these two functions as Gibbs energy and Helmholtz energy, respectively; several other terms are also used for them. The Helmholtz free energy is considered as a thermodynamic potential which calculates the useful work obtainable from a closed thermodynamic system at a constant temperature and volume. For such a system, the negative of the difference in the Helmholtz energy (-ΔA) is equal to the maximum amount of work extractable from a thermodynamic process in which both temperature and volume are kept constant. A = U-TSΔA = ΔU –TΔS (as ΔT = 0)or ΔA = ΔU – q…………(12) (as ΔS = q/T )Now, from first law of thermodynamics, we getΔU = q + w or w = ΔU – q Putting it in equation (12)ΔA = wor ΔA = -wmaxmax (work done by the system = -w)or -ΔA = wmax6.17.1 Relationship between Gibbs free energy (G) and Helmholtz free energy (A)The two functions can be defined as G = H - TS and A = U – TSAt constant temperatureΔG = ΔH – TΔS ΔG = (ΔU + PΔV) – TΔS (as ΔH = ΔU + PΔV)ΔG = (ΔU – TΔS) + PΔV (as ΔA = ΔU – TΔS at constant temperature)ΔG = ΔA + PΔV ΔG = -wmax + PΔV (as ΔA = -wmax)-ΔG = wmax - PΔV 6.18 Third law of ThermodynamicsAs the temperature of any system decreases, the atom and molecules in the system lose their energy and approach their lowest energy points, by doing so atom and molecule’s kinetic energy decreases and hence their random motion. For example, the water molecules in the ice crystal are highly ordered and entropy of the system is very low. If the solid crystal is further cooled, the vibration of molecules held in the crystal lattice gets slower and they have very little disorder and hence very small entropy and finally at absolute zero (0 K or − 273 ºC), theoretically all modes of motion stop.
Absolute zero is a temperature that an object can get arbitrarily close to but is not attainable and a perfect single crystal is also an ideal that cannot be achieved. So, when absolute zero temperature is achieved, the atom and molecules of the crystal have the lowest energy and hence no randomness in the particles leading to zero entropy of the system. The third Law states, “the entropy of a perfect crystal is zero at a 0 K temperature or called absolute zero”. This is so because there is perfect order in a crystal at absolute zero. 6.18.1 Application of Third LawThird law allows us to measure absolute values of entropy (S) for a substance at 0 K from thermal data alone. Unlike enthalpy or internal energy, it is possible to obtain absolute entropy values by measuring the entropy change that occurs between the reference point of 0 K (corresponding to S=0). For a pure substance, this can be done by calculating the increments of heat q required to bring the substance from 0 K to the temperature of interest, and then summing the ratios q/T. Crystals with defects (imperfection) at absolute zero, have entropy greater than zero. Absolute entropy of a substance can never be negative.
Subscribe
Would you like to receive our Newsletter, NEET- IIT Videos, STEM Materials, NEET-IIT Solved previous year question papers.
Book Your Demo Class
Try for 1 Month @
Rs 2,000 only few seats available. Offer Ends Soon.
Book now and get your free unlimited TeenEinstein Portal Access.
You are now subscribed to our Newsletter etc.
Would you like to continue Free Trial.
TeenEinstein Stem Platform Portal access for 30 days Would you like to get the Portal access free trial for 30 days .
TakeBreak
You have watched Screen for 30 minutes, please relax your eyes by watching any other object at 20 feet for 20 secs.